EnsDeconv
EnsDeconv.RmdInstall packages
# install devtools if necessary
if (!"devtools" %in% rownames(installed.packages())) {
install.packages('devtools')
}
# install the EnsDeconv package
if (!"EnsDeconv" %in% rownames(installed.packages())) {
devtools::install_github("randel/EnsDeconv", dependencies = T)
}Sample Code
Use two reference data, one deconvoluton methods, one normaliztaion, one transformation, one marker gene approach.
The testdata includes two different reference dataset (Nowakowski and Darmanis), and sample bulk data (n = 10).
## Warning: package 'scran' was built under R version 4.2.2## Warning: package 'SingleCellExperiment' was built under R version 4.2.2## Warning: package 'S4Vectors' was built under R version 4.2.2## Warning: package 'GenomeInfoDb' was built under R version 4.2.2## Warning: package 'scuttle' was built under R version 4.2.2
data(testdata)
params = get_params(data_type = "singlecell-rna", data_name = names(testdata$ref_list), n_markers = 50, Marker.Method = "t", TNormalization = "none", CNormalization = "none", dmeths = "CIBERSORT")
res = EnsDeconv(count_bulk = as.matrix(testdata$count_bulk), ref_list = testdata$ref_list, params = params)
# Use parallel computing
# res = EnsDeconv(count_bulk = as.matrix(testdata$count_bulk), ref_list = testdata$ref_list, ncore = 4, parallel_comp = T, params = params, outpath= '...')
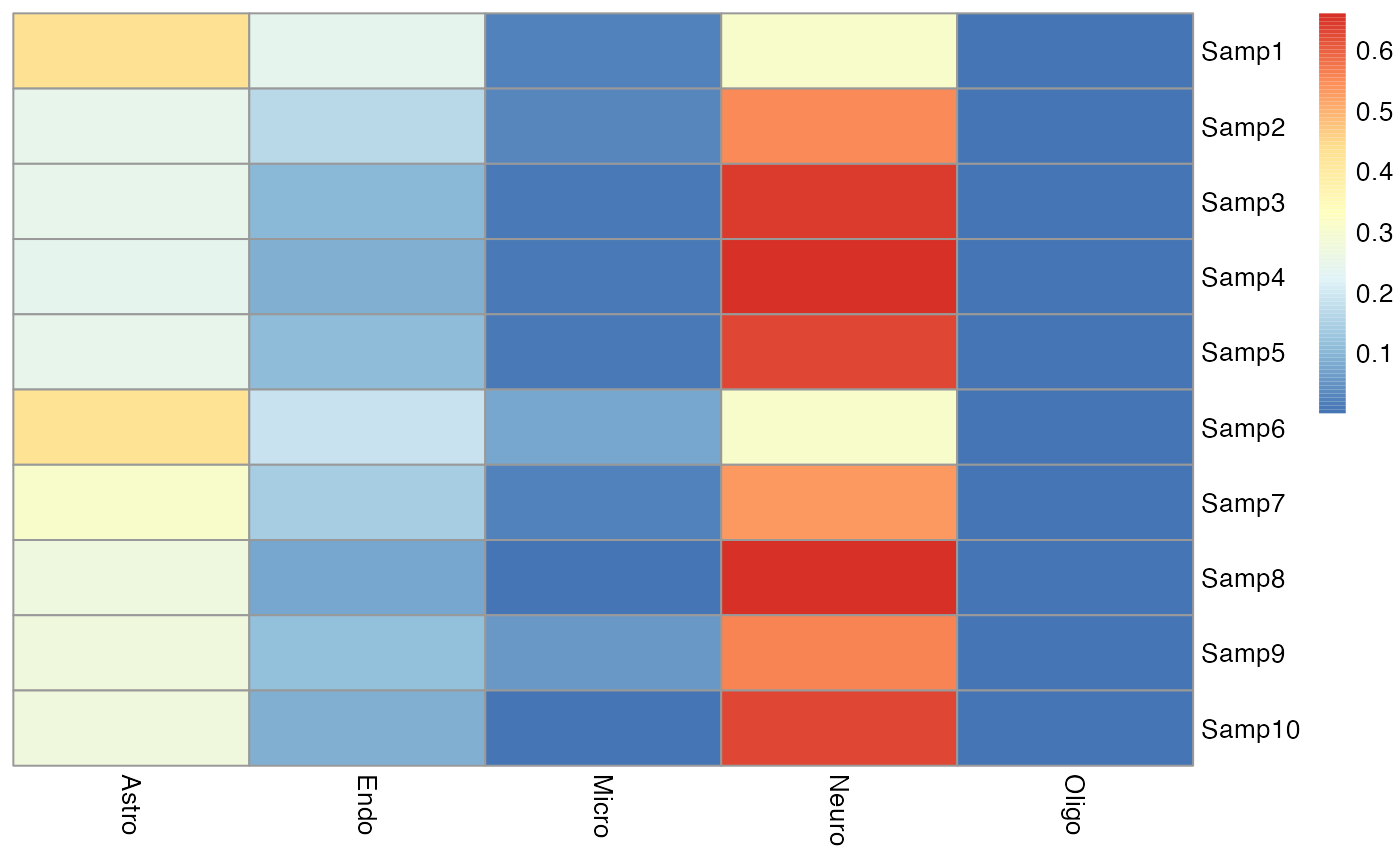
pheatmap::pheatmap(res[["EnsDeconv"]][["ensemble_p"]],cluster_rows = F, cluster_cols = F)