bMIND: Bayesian estimation of cell-type-specific (CTS) gene expression and CTS differential expression analysis
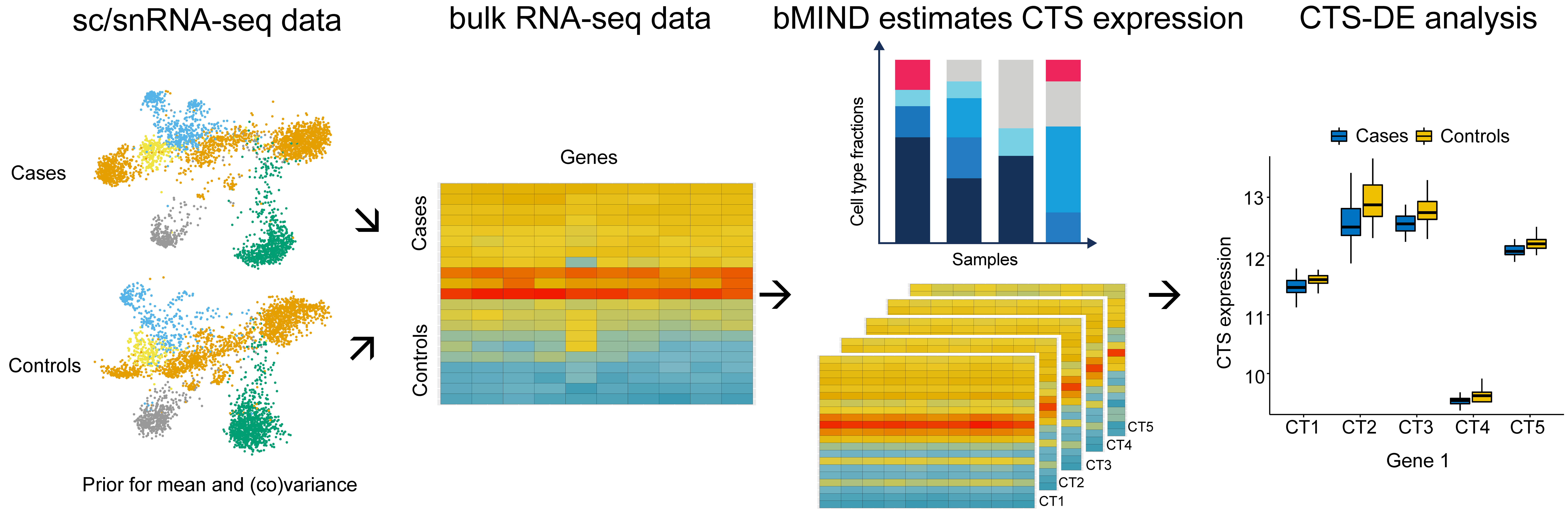
bMIND is a Bayesian deconvolution method to integrate bulk and scRNA-seq data. With a prior derived from scRNA-seq data, we estimate cell-type-specific (CTS) expression from bulk tissue expression via MCMC.
Example
library(MIND)
data(example)
bulk = t(na.omit(apply(example$X, 1, as.vector)))
frac = na.omit(apply(example$W, 3, as.vector))
colnames(bulk) = rownames(frac) = 1:nrow(frac)
y = rbinom(n = nrow(frac), size = 1, prob = 0.5)
covariate = data.frame(c1 = rnorm(length(y)), c2 = rnorm(length(y)))
# CTS-DE (np = TRUE: use non-informative prior)
deconv = bmind_de(bulk, frac, y = y, covariate = covariate, covariate_bulk = 'c1', covariate_cts = 'c2', np = T)
# estimate CTS expression
deconv2 = bMIND(bulk, frac)Tutorials
For detailes, please see the tutorial. It covers how to get prior distributions using multi-sample scRNA-seq data.
The cell type fraction can be pre-estimated using 1) non-negative least squares (NNLS), which requires a signature matrix derived from reference samples of single-cell RNA-seq data; 2) Bisque, which requires raw single-cell data.
Reference
bMIND: Wang, Jiebiao, Kathryn Roeder, and Bernie Devlin. “Bayesian estimation of cell type-specific gene expression with prior derived from single-cell data.” Genome Research (2021) 31: 1807-1818.
MIND (frequentist method): see https://github.com/randel/MIND/blob/master/MIND.md
Common errors
If you see strange error, first check the dimnames and each cell type should have an average fraction > 5%. The Bayesian software does not allow dots in the bulk sample name. Try colnames(bulk) = rownames(frac) = paste0('s', 1:nrow(frac))
Also remember to remove any dot/space in the first cell type name, e.g., colnames(frac) = paste0('c', 1:ncol(frac))